Aliyu Mahmuda1,2,
Faruku Bande3,
Khalid Jameel Kadhim Al-Zihiry4,
Noor Abdulhaleem5,
Roslaini Abd Majid1,
Rukman Awang Hamat1,
Wan Omar Abdullah6,
Zasmy Unyah1 ![]()
For correspondence:- Zasmy Unyah Email: ngah@upm.edu.my Tel:+60132213516
Received: 11 November 2016 Accepted: 19 February 2017 Published: 31 March 2017
Citation: Mahmuda A, Bande F, Al-Zihiry KJ, Abdulhaleem N, Majid RA, Hamat RA, et al. Monoclonal antibodies: A review of therapeutic applications and future prospects. Trop J Pharm Res 2017; 16(3):713-722 doi: 10.4314/tjpr.v16i3.29
© 2017 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
The increasing demand for monoclonal antibodies (mAbs) used for diagnostic and therapeutic applications has led to the development of large scale manufacturing processes, with improvements in production achieved through continuous optimization of the inherent systems. The number of monoclonal antibodies (mAbs) that have already been approved for therapeutic applications and for use in clinical trials have significantly increased in the past few years. In view of the side effects and limitations of mAbs, several improvements and modifications to monoclonal antibodies have been developed. These modifications have facilitated the use of mAbs in various forms of therapeutic applications such as treatment of infectious diseases caused by bacterial, viral, fungal and parasitic organisms. Monoclonal antibodies have also been applied in the treatment of non-infectious diseases such as cancer, immune diseases, arthritis and other disorders resulting from organ transplantation. This review highlights mAbs applications in biomedicine, and discusses state-of-the-art technologies related to their potential uses
Introduction
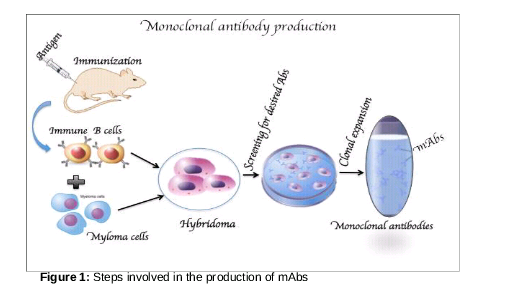
The immune system acts as defence against various infectious agents that cause different forms of diseases. Two major components are the humoral (antibody-mediated) and cellular (cell-mediated) immune responses. The humoral immune system which comprises B-lymphocytes recognizes the type of foreign invading antigens and produces specific antibodies against them [1]. The two important characteristics of an antibody are its specificity to the antigen, and its assurance to provide continual resistance to that particular type of antigen [2]. Considering their unique features, scientists use them for the protection of humans against diseases. Techniques for in vitro production of antibodies was also developed, resulting in the production of monoclonal antibodies for diagnostic and therapeutic applications [3]. Although antibody production has several procedures, the principles generally remain similar to those illustrated in .
Due to development of new phase of therapy in the field of medicine, application of mAbs in the treatment of several disease conditions have been at the forefront [4]. In 2002, the first human mAb for use in clinical practice was approved by the Food and Drug Administration of the United States. Since then, the mAbs production industry has exponentially expanded [5]. The use for which mAbs are anticipated determines the exact amount required for carrying out the different activities. Only a small amounts of mAb (0.1 g) is required for carrying out most research and analytic work [6].
About 30 mAbs were recently accepted for clinical use as therapeutics, with several others been at various trial stages [7]. Although there are current studies that are aimed at improving the efficacy of existing mAbs through optimisation processes, there still exist some limitations one of which include lack of efficient mAb generation models [8]. However, despite these shortcomings, there remains to exist an interest by pharmaceutical companies to develop mAbs for clinical use. This is expected to control the management of many diseases in future base on both clinical and economical perspectives [9].
Types of therapeutic MAbs
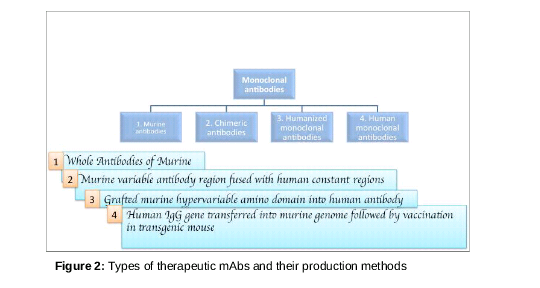
Progress in antibody engineering has yielded various types of mAbs for application in life science and biomedicine. These types of antiboies may have similar principles, but different targets and applications. In addition, the choice of one method over another may be guided by several factors, including purpose of application, availability and effectiveness ().
Murine MAbs
The use of murine antibodies produced by hybridoma technology in human therapy (clinical medicine) is limited, attributable to differences between the human and rodent immune systems. This usually results in treatment failure, with the exception of some particular circumstances [10] . Murine antibodies have mild effects of stimulation of cytotoxicity. Thus, their continuous administration often results in allergic reactions and anaphylactic shock, a result of the production of human anti-mouse antibodies (HAMA) that invariably attack the administered murine mAb and in turn stimulate allergic response [6-8]. Anti-CD3 mAb of murine origin (OKT-3) was the first therapeutic mAb that was approved for clinical use in human medicine. However, the mAb failed in treatment of transplantation rejection, primarily because it causes severe human anti-mouse antibody (HAMA) response in patients [11]. In order to minimize immunogenic effects of murine mAbs in human therapy, murine immunogenic components are removed with increased efficiency through various approaches [12].
Since murine mAbs contain foreign protein molecules, majority of the early reagents for clinical use stimulated unwanted immune responses in human patients. Recently, molecular biology advances have brought about in vitro gene manipulation and subsequent expression of these manipulated sequences in mammalian, bacterial or fungal cell culture protocols. This has thus ensured a better option for re-engineering murine mAb to partly substitute the rodent antibody fragment with a corresponding human antibody sequence.
The total immunogenicity of the mAb is therefore reduced without affecting the recognition ability of the original antibody [13]. Antibodies resulting from humanisation are becoming more relevant in the treatment of inflammatory diseases and cancer, with several antibody products readily available in the market, and others undergoing clinical trials [14].
Chimeric MAbs
Chimeric antibodies are special types of therapeutic antibodies made by the combination of genetic ingredients from humans and non-humans (mice). They are produced through manipulation of human constant regions and mouse variable regions [15]. These antibodies are made up of about 65% human genetic component in order to minimize the risk of unwanted reactions to foreign antibodies. Interestingly, the Food and Drug administration has approved some drugs that are based on chimeric antibodies for use in human therapy and research. The nomenclature for naming chimeric mAbs ends with the suffix “ximab” e.g., Infliximab, Rituximab, Abciximab [16].
Humanised MAbs
Human mAbs (HMA) have been considered natural drugs due to their safety for in vivo activities. Modifications in the field of mAb technologies has made human mAbs to be widely applied in the therapy of various diseases, as well as in the development of novel immunodiagnostics. A number of about 20 mAb-drugs, including humanized mice mAbs, have been accepted as therapeutic reagents during the past few decades. Other mAbs at different stages of clinical trial, and controlled by different research institution, and/or in collaboration with pharmaceutical companies. Use of human mAb technologies are not only limited to strategic research, but are also of great values in health economics [17].
In humanised antibodies, the hyper variable regions are grafted onto human variable domain framework. The antibody molecules is nearly 95% human origin. They are sometimes weaker than the parent murine monoclonal antibodies in terms of binding with antigens [23]. To increase the antibody-antigen binding affinity, techniques such as chain-shuffling randomisation can be employed to introduce some transformations into the complementarity determining region (CDR). Examples of FDA-approved humanised antibodies include daclizumab, omalizumab, alemtuzumab [18].
Fully human MAbs
Human mAb production by the conventional hybridoma techniques is relatively difficult because of the stress involved in maintaining immortalised cell lines and human hybridomas. It is also not feasible for in vivo immunization of humans with many different antigens compared to the use of animal models [10]. However, methods for the production of human mAbs are made possible through the expression of antibody fragments or single cell variable fragment (Fab or ScFv) in bacteria. Similarly, antibody fragments can be displayed on filamentous bacteriophages for screening of antibody libraries [19,20]. Generation of fully human mAbs serves as an alternative to re-engineer murine mAbs with a source of low immunogenic therapeutic antibodies. Most of these drugs were generated using either transgenic mice or phage display platforms. However, there is still no clear distinction between them.
The phage display technique is a well-established and the most widely used method for the development of new human antibodies [21]. Alternatively, using transgenic mice containing human immunoglobulins may be a strategy for the production of human mAbs. Hybridomas that produce human antibodies can be generated due to a human antibody response resulting from the immunization of transgenic mice.
In 2003, Humira®, the first fully human mAb drug was lunched for the treatment of rheumatoid arthritis [22]. Adalimumab® and Panitimumab® are among the marketed fully human therapeutic mAbs, while several others are in various stages of human clinical testing. Two basic platforms have demonstrated to yield active and well-tolerated therapeutics for the clinical use of fully human mAbs. These include transgenic mice and phage display platforms [23].
Transgenic mice platform
Mice that express human antibody repertoires through genetic engineering were first reported for developing human mAbs using conventional cell culture technique [17]. The manipulation of endogenous mouse immunoglobulin heavy chain and immunoglobulin light chain loci is the result of the transchromosonal engineering process. Transgenes encrypting human immunoglobulin heavy and light chains were introduced. Progress has been achieved to express multiple Vgene segments by transgenic mice over the past few decades, with increase in the number of potential repertoires of the recovered mAbs [25].
A generation of transgenic mice producing human mAbs with various heavy chain isotopes was achieved; although immune responses are sometimes less robust as observed in transgenic mice compared to those observed in normal mouse strains used in generating mouse mAbs. In spite of the above limitations, about six different human mAbs produced by transgenesis have been approved for marketing and over 50 human mAbs in clinical trials [17]. This expression system of human immunoglobulin in transgenic mice prevents the unwanted human anti-mouse antibody responses, and maintains the merits of murine conventional cell culture technique for the generation of potential therapeutic reagents [30].
Phage display platform
The phage display library technology was first established in 1985 and has been employed to generate a large number of short peptides and protein molecules on bacteriophages. This allowed the cloning of foreign DNA sequences into filamentous bacteriophage and subsequent expression of the clones on the surface of the phage particles as fusion proteins [24]. This technology has been effectively used to select antigen-specific variable region genes as well as to express purposeful and specific antigen fragments [21]. The phage display approach has been combined with PCR means to clone expressed immunoglobulin variable region cDNA repertoires so as to produce diverse libraries of phage displayed antibody variable region. This could be applied to promptly access target-specific mAbs, without creating a library of hybrid clones [25]. Reverse transcription of messenger RNA (mRNA) from B-cells and PCR amplification methods were also used to isolate human antibodies. Using this same technology, a library of different human immunoglobulin heavy chain variable (VH) genes and light chain variable (VL) gene segments were produced [21].
The process of combined infection and in vivo recombination has been used to display single chain fragment variable (scFV) on the surface of the phage, to generate large antibody repertoires [26]. Identification of specific antibody fragments that have good affinities is possible after bio-panning the phages. The purpose of this is to increase the efficacy of antigen-specific scFv, in order to enhance the affinity of scFV for antigens, and with increased specificity [27]. Similarly, phage display technology has been improved, and currently, about 35 human mAbs through this technology have been certified as therapeutic reagents by the Food and Drug Administration [22].
Therapeutic applications of MAbs
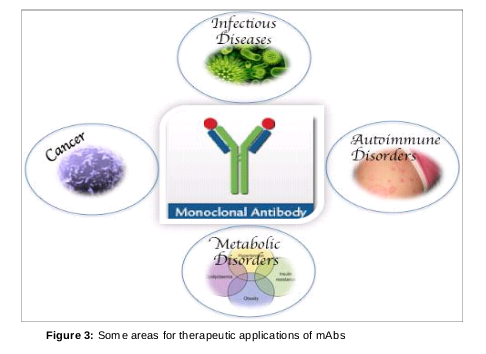
Recent advances in genetic engineering have made possible efforts to improve the therapeutic application of mAbs by identifying new targets with improved efficacy for use in clinical practice [28]. Their use in immunoprophylaxis or immunotherapeutics have been extensively applied to infectious diseases, as carriers for toxic substances delivery to tumors or as tools for identifying, locating and target neoplasms [29]. They have also been used in the treatment of several types of cancers, immune diseases, arthritis and metabolic diseases (). Their therapeutic applications include cancer therapy, human and animal disease therapy, preparation of vaccines, suppression of immune response and purification of hormones [30].
Infectious diseases
Researchers have shown that mAbs could potentially prevent effective colonization of Streptococcus mutans in cases of dental caries. With the identification of new peptide sub-units (epitopes) of Streptococcus mutans, mAbs could be used to effectively treat these conditions. In this regard, the endogenous bacteria (Streptococcus spp, Lactobacillus spp) are colonised to form the main antigen [31]. The mucosal defence mechanism is mostly mediated by secretory antibodies (sIgA) of the saliva. Immunization (vaccination) with purified antigen of Streptococcus mutans helps in mobilizing antigen-specific immunoglobulins, particularly IgA, to salivary glands at induction sites. These immunoglobulins (IgA) are produced by specific B-cells due to their differentiation and maturation in the saliva. Some non-human sources of mAbs have also been developed with a lack of side effects such as allergic reactions [6].
Neutralizing antibodies against highly variable viral pathogens protects against globally circulating viruses [32]. Probing of the neutralizing antibody repertoires of HIV donors with unusually extensive and effective responses yielded 17 new monoclonal antibodies. Many of the new mAbs are nearly ten-fold more potent compared to the recently described PG9, PG16 and VRC01 mAbs; and 100-fold more potent compared to the original prototype HIV broadly neutralizing mAbs [32].These mAbs have recognised novel epitopes on envelop glycoprotein (gp120), thus revealing new alternative targets for vaccine design. Analysis of the neutralization by these available mAbs reveals that certain mixtures of antibodies could offer a more favourable and diverse coverage of circulating viruses [32].
Since mAbs that specifically targets the C9 binding site of Trichinella spiralis paramyosin (Ts-Pmy) would be necessary for use in the treatment of Trichinella spiralis infection, mAb 9G3 was successfully produced. It was found to be reactive to recombinant Ts-Pmy, as well as to native Ts-Pmy expressed in different stages of Trichinella spiralis. The binding of the mAb (9G3) to Ts-Pmy was found to actively antagonise the binding of Ts-Pmy to human complement C9, thus leading to a significant increase in the killing of new born larvae in vitro, and a reduced infectivity of Trichinella spiralis in the mAb-treated mice [33]. Conclusion was drawn that the mAb (9G3) is both a preventive and therapeutic reagent for Trichinella spiralis infections.
Cancer therapy
Monoclonal antibody mediated therapy recruits cytotoxic cells (monocytes and macrophages) through antibody-dependent cell cytotoxicity [34]. In the treatment of cancer, mAbs binds complement proteins, which thus lead to direct cell cytotoxicity that is complement-dependent in nature [3].
Some monoclonal antibodies have been reported to function in blocking the growth factor by binding to, and inhibiting, growth factor receptors, in order to effectively arrest the proliferation of the tumor cells [42]. Ibritumomab is a monoclonal antibody against the CD20 antigen on B-cells for the treatment of lymphoma patients; while Rituximab (IDEC-C2B8) is a chimeric immunoglobulin (IgG) mAb directed against the CD20 antigen, which has been reported to be effective against B-cells malignancies [22].
Monoclonal antibodies are also modified for the delivery of radioisotope (Radioimmunotherapy), toxins, cytokines and several other active conjugates. Bi-specific mAbs can be designed so that their Fab region can target both antigen and the effector cells. Report from a study have also shown that conjugates, toxins and drugs with mAbs can also kill leukemic cells [35]. Cetuximab® is a drug used in the treatment of certain types of breast cancers and lymphomas by blocking HER-1; while Trastuzumab® is known to block HER-2 receptors in breast cancer [36]. Therapeutic anti-cancer mAbs against leukemia are Gemtuzumab® and Alemtuzumab®; while Nimotuzumab® and Cituximab® are for carcinomas. Bevacizumab is another is another drug (mAb) approved by the Food and Drug Administration (FDA) for use in the therapy of colorectal cancers. It binds to vascular endothelial growth factor (VEGF), thereby preventing it from binding to its receptors. Vitaxin® is a clinical trial (phase II) drug that yielded a better results in shrinking solid tumors, with no associated detrimental side effects. It is found to binds to a vascular integrin (alpha-v/beta-3) present in blood vessels supplying tumors but is absent in blood vessels found in normal tissues [37].
Auto-immune diseases
Infliximab® and Adalimumab® are available monoclonal antibodies used for immune diseases, and are effective against rheumatoid arthritis, Crohn’s disease and ulcerative colitis [6]. They have the tendency to bind and block tumor necrosis factor (TNF), TNF-α, and Interlukin-2 (IL-2) on activated T-cells which are also inhibited by Basiliximab and Daclizumab, and thus help prevent acute rejection of kidney transplants. Daclizumab® is also a potent mAb-drug for the treatment of T-cell lymphoma while inhibition of human IgE by Omalizumab® is valuable in the treatment of various types of allergic asthma [38]. OKT3 (Muromonab®, Orthoclone®) is the first FDA approved therapeutic mAbs (murine IgG2a; CD3-specifc), and is currently used in steroid-resistant patients who suffer from rejection after solid organ transplant. Patients that have received kidney transplant are routinely given this drug to induce immunosuppression, in order to prevent rejection of the foreign tissue. The (OKT-3) mAb is known to attack the T-cells that cause rejection [37].
In attempts to determine whether monoclonal antibodies directed against immune cell components responsible for eliciting abnormal immune response could be used to treat such conditions [39]. Monoclonal antibodies against the endotoxins of Escherichia coli have been produced and have protected mice from bacteremia. They have also been analyzed in humans. An anti-T-cell monoclonal antibody that remove T-cells from marrow of a donor prior to transplantation, is also available which leads to reduction in graft-host diseases reduction in graft-host disease [40].
Metabolic disorders
There is no doubt that metabolic disorders such as diabetes, hypercholesterolemia have posed a great challenge in human medicine. One of the areas where mAbs therapeutics is applied includes metabolic disorders. G protein-coupled receptors (GPCRs) are implicated in a wide variety of metabolic diseases. Thus, scientist have used GPCRs membrane fractions as a target to produce mAbs for the cure of metabolic disorders [41]. Monoclonal antibodies have been produced against the human glucagon receptor (GCGR) from stable cell lines through a transgenic XenoMouse platform. These candidate mAbs were shown to display potential antagonistic activity in reducing blood glucose level with a resultant long-term inhibition of GCGR signalling in a mouse model, making them effective for controlling diabetic hyperglycemia [43].
Scientists at Harvard School of Public Health have recently discovered a new treatment for type 2 diabetes through the use of novel mAbs targeting secreted fatty acid–binding protein aP2. This is an intracellular chaperon protein (aP2FABP4) that contributes to hyperglycemia by either encouraging hepatic gluconeogenesis or countering peripheral insulin action, or by combination of both. Fatty acid-binding proteins (aP2) have been implicated as a cause of pathology of many metabolic diseases in humans. Following the experimental treatment of mice with aP2-mAb, there was enhanced total glucose metabolism, elevated systemic insulin sensitivity, lowered fasting blood glucose and reduced fat mass and liver steatosis in obese mice models. This approach has open a promising paradigm for the treatment of type 2 diabetes [43].
Among the major risk factors for cardiovascular diseases is hypercholesterolemia. The effective management of this condition has mainly been achieved by statins as first-line therapy, with a reported reduction of cholesterol level up to 50 % have been reported [44]. Others are recommended only if statins are not tolerated. Diet-based management is the mainstay of therapy in children. However, as of 2010, no study has shown improved clinical outcomes [45]. This necessitates the use of other lipid-lowering agents due to adverse effects in statins intolerable patients, especially those with very high risk of cardiovascular diseases [46]. Increase low-density lipoprotein cholesterol (LDL-C) levels have been reported to be a major cause of metabolic disorders. Studies that aims to inhibit the proprotein convertase (subtilisin/kexin) type 9 (PCSK9) to lower LDL-C levels have been underway through the development of mAbs. Evolocumab and Alirocumab were found to be safe and well-tolerated [47]. Both antibodies were reported to substantially reduced the LDL-C level by over 50%, increase high-density lipoprotein cholesterol (HDL-C) level and result in favourable changes in other lipids [48].
Problems and future prospects of monoclonal antibody therapy
The concept that all therapeutic agents have unwanted effects is also applicable to monoclonal antibodies. These range from mild to severe signs depending on the class they belong to, and the route through which they are administered [49]. A mild allergic reaction (rash) may appear with the first administration of mAbs. Clinical signs of weakness, headache, fever, diarrhoea, vomiting and nausea, and sometimes decreased blood pressure, are common side effects. A mAb used against tumor blood vessel growth (Bevacizumab®) can present numerous side effects, some of which include renal failure, bleeding with poor wound healing and high blood pressure. Injection with FDA approved mAb (Raxibazumab®) for the treatment of infectious inhalational anthrax is also associated with rash and severe itching, extreme pain and drowsiness [50].
Only a few out of many FDA regulated mAbs are available in the market, while a large number of new mAb drugs under development [51]. These drugs have always been very expensive and almost un-affordable by patients. In the absence of competition (generic), the sale of the first generation mAbs is quite good in terms of safety and price. However, with the high demand of specialty pharmaceuticals, steps have been taken to transform the manner in which mAbs are produced, commercialised and marketed. This has made mAb drug therapies a financial burden on patients, requiring some eminent health plans and step-wise therapies to resolve [52]. Some mAb drugs (e.g Bevacizumab) after repeated use were found to be widely available and cheaper than other agents (e.g Ranibizumab®), while demonstrating similar efficacy. This is because of the competition between improved mAb and a previously established product, which requires a comparative clinical assessment for justification [53].
Future prospects of monoclonal antibodies lie in improvements and beneficial modifications of therapeutic mAbs, so as to reduce their harmful effects as antibody-based drugs. Monoclonal antibodies can also be further modified in order to have improved effects, through the possibilities of conjugated antibodies with coupling effector molecules [54]. The majority of mAbs from the early transgenic mice platform still remain murine in nature. These strains, despite their profound defects in B-cell development, they also remain to contain the necessary factors for immunoglobulin gene function, transcription and immunoglobulin gene recombination [55]. Chimeric antibodies, on the other hand, despite their limitation for therapeutic purpose, they are useful standardization reagents in diagnostic assays [56]. The XenoMouse strains were the first engineered mice with a majority of both human VH and VK repertoires, causing them to offer superior genetic stability [25]. This is seen as a benefit in design of potential therapeutic antibody with or without the effector function. About five fully human mAbs from XenoMouse strains have been used in human therapy [57].
Efforts to minimize non-human sequences of therapeutic mAbs involve immunogenicity, such as that seen in antibody fragments, which are usually less immunogenic due to their lack of Fc domain. In order to increase mAbs efficacy, the treatment of antibody fragments with polyethylene glycol (PEG) has also been applied [58]. The plasma half-life of IgG isotypes have also been reported to increase by developing antibodies through phage display platforms, and with noticeable increased affinity [59]. Binding affinity of an antigen can be up-graded through phage display libraries to isolate antibodies with strong affinities for the antigen, which consequently improves their therapeutic potentials [60].
Conclusion
Although the first mAbs approved as human therapeutic agent were generally reported to be intolerable as therapeutics, advances in hybridoma technology have resulted in mAbs that are currently more effective and safe. Many of these new generation mAbs have been approved for human therapy. Multiple engineering efforts have led to the evolution of modified therapeutic antibodies in the hope of improving their efficacy and safety as antibody-based drugs. These efforts include antibody chimerization, humanization and the development of fully human antibodies. The ability to engineer variable regions that encode multiple specificities into a single molecular entity has been an advantage for optimizing antigen-binding capabilities. There is no doubt that the future of mAbs will sway the treatment of infectious diseases, cancers and other conditions like Alzheimer and Parkinsonism. Whether therapeutic mAbs will be commercially more abundant and affordable in the near future, is in part, a matter of rapid advances in biotechnology and biomolecular sciences as well as the outcomes of extensive clinical trials.
Declarations
Acknowledgement
References
Archives


News Updates